




















蛋白质是生命活动的主要承担者,肌肉的收缩、食物的代谢消化、血液内氧气的运输、抵御细菌的入侵……所有的这些都离不开蛋白质的参与,甚至毫不夸张的说,没有蛋白质就没有生命。自然界内蛋白质千奇百怪,功能各异,然而它们都起源于同样的基础结构,一条由20 种不同氨基酸串成的线性链。在被称为核糖体的细胞工厂中组装后,每条链折叠成独特的、精致复杂的3D结构。正是这些三维结构决定了蛋白质的功能和在细胞内的作用,决定了蛋白质如何与其他蛋白质、DNA、RNA等分子相互作用。
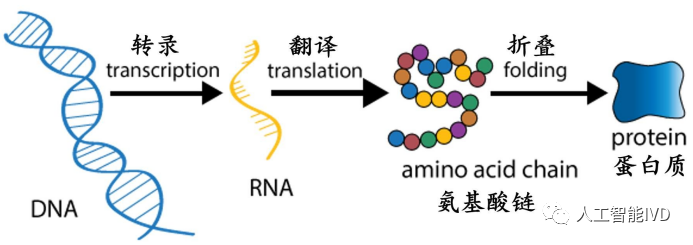
中心法则(图源:[1])
蛋白质的氨基酸序列由我们的DNA所编码,DNA转录为RNA,再翻译为蛋白质,从而实现生命功能,而蛋白质必须正确地折叠成三维结构,才能发挥其功能。
五十年前,Christian Anfinsen提出了关于蛋白折叠的猜想:蛋白质的天然结构是一个热力学稳定的状态,可以基于其氨基酸系列来预测蛋白质如何折叠成天然结构。然而,蛋白质的结构复杂性远超人们的想象。一个蛋白质分子是由一条或几条多肽链组成,多肽链则折叠成特有的形状。同时,蛋白质分子的专一形状是由4个层次的结构决定的,包括一级、二级、三级和四级结构,前一级结构决定后一级结构。其中多肽链的氨基酸序列是一级结构,一级结构中的部分肽链卷曲或折叠,多肽链上氨基酸残基相互之间形成氢键、静电相互作用、疏水相互作用,产生二级结构,主要包括α螺旋和β折叠等。二级结构经过一系列的构象改变形成三维结构即三级结构,一般为球状或纤维状。三级结构有特定的结构区域,形成结合口袋或位点,可以结合特定结构的生物活性物质,行使蛋白的功能。两条或两条以上的多肽链组成的蛋白质,可以形成四级结构。
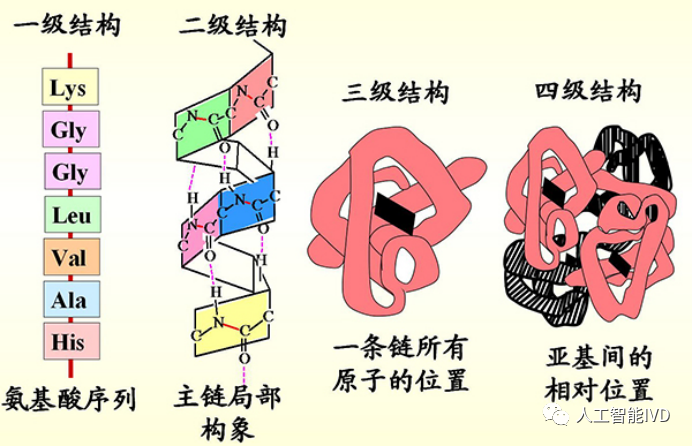
蛋白多级结构层次(图源:网络)
常规研究蛋白质折叠的方法主要为借助实验仪器解析蛋白质结构如:X-射线衍射、核磁共振技术、冷冻电镜技术等,然而这些实验技术都有着很大的局限性。X-射线衍射对蛋白质纯度要求很高,并非所有的蛋白质都能获得高纯度的晶体用于衍射实验。近年来,随着冷冻电子显微镜技术的发展,可以在没有结晶样本的条件下观察蛋白质结构,使得蛋白质结构研究有所进展。然而,冷冻电镜是非常昂贵的设备,只有极少数的实验室才有条件配备,也需要复杂耗时的实验流程操作和结果解析,对于广大科研工作者非常不友好。因此,从Christian Anfinsen猜想提出至今 50 多年的时间里,科学家始终无法解决蛋白质折叠的问题,对于蛋白质结构的了解依旧十分有限。
随着计算机科学的发展,此前曾有学者提出利用计算机模型解决蛋白质折叠问题。虽然这一想法是可行的,但是在随后数十年的时间里,人们开发的各种计算机模型预测蛋白结构的准确性始终有限。3D结构在形成之前会有数以亿计的折叠方式。假设一条含有100个氨基酸的多肽/蛋白质,其每个氨基酸残基只能处于α螺旋、β折叠或无规线团三种状态,那么整条肽链拥有3100个状态,约为5*1047,根本无法计算如此天文数字所包含的所有可能构型。在过去25年中,国际蛋白质结构预测大赛Critical Assessment of protein Structure Prediction (CASP)一直关注这个领域的进展,试图寻找能够完美解决蛋白质折叠问题的计算机模型。大赛组织者提供多个蛋白质的氨基酸序列,这些蛋白质的结构已经通过实验方法被解析出来,但还没公布。参赛人员利用开发的计算机模型对蛋白质最终折叠成的结构进行预测,最后将预测的结构与实验获得的结构进行比对评分,高于90分则认为与实验方法解析的结构接近。
早期的计算机预测结果与实验结果偏差非常大,中位数得分低于60。到2018年,大部分的计算机模型得分也不过是在75分左右。而直到2020年第 14 届大赛CASP14大赛成功举办,谷歌旗下人工智能公司DeepMind的AlphaFold系统才在蛋白质结构预测方面表现出了无与伦比的准确性,取得了中位数得分92.4(满分100分),比第二名高25分,打败了所有竞争对手。即使是在最难的没有同源模板的蛋白质上面,这个分数也达到了恐怖的87.0。同时,AlphaFold 的神经网络能在几分钟内预测出一个典型蛋白质的结构,还能预测较大蛋白质(比如一个含有 2180 个氨基酸、无同源结构的蛋白质)的结构。该模型能根据每个氨基酸对其预测可靠性进行精确预估,方便研究人员使用其预测结果。
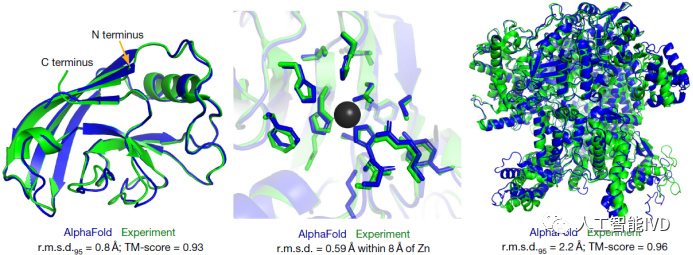
AlphaFold预测结构与实验结构的对比(图源:[2])
而在AlphaFold之后,在2021年7 月份,华盛顿大学医学院生物化学系教授、蛋白质设计研究所所长 David Baker 领导一支计算生物学家团队,成功开发一款名为 RoseTTAFold 的工具,基于深度学习,能够根据有限的信息快速准确地预测出目标蛋白质的结构,达到与 AlphaFold2 不相上下的准确度。之后DeepMind 公司与欧洲分子生物学实验室(EMBL)下属的欧洲生物信息学研究所(EBI)合作,公布AlphaFold-EBI蛋白结构库,利用 AlphaFold2,进一步对整个人类蛋白质组98.5%的(20296种)人类蛋白的结构做出了预测。此外,该工具预测了各种其他生物的几乎完整的蛋白质组,从小鼠和玉米到疟疾寄生虫,预测的蛋白结构总数多达35万个,并且预计在2022年将完成1.3亿条蛋白质序列的结构预测。这次预测的蛋白质结构保存在一个公用数据库中,虽然这些预测的准确度各不相同有高有低,但研究人员表示这些资源将有可能彻底改变生命科学。
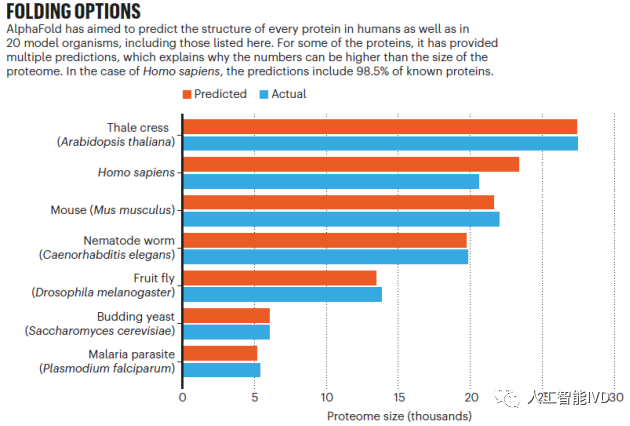
AlphaFold预测了智人和20种模式生物的逾35万个结构(图源:[3])
蛋白质的结构决定了它的功能,因此揭示蛋白质的结构可以帮助我们理解蛋白质的功能。如果我们清楚蛋白质的结构,就能对其功能展开有根据的猜测。通过绘制大量蛋白质结构图,我们可以解读生命的生物学原理,并找到解决问题的根本方法,而结构解析目前远远超出了人类的认知能力。也正因如此,很多实验生物学家将大量时间、精力投入到蛋白质结构实验解析上,也正因为这一进程发展缓慢且困难重重,才促使人们另辟蹊径,借助计算科学开展结构预测。在这个意义上说,实验解析或算法预测是开展蛋白质功能研究及认识生命机理的工具和手段。AI预测蛋白质结构的成功为我们攻克诸多重大疾病提供了助力。我们设计的几乎所有的药物都作用于蛋白质,如钥匙开锁般精确匹配,而此过程的第一步是确定哪把钥匙开哪把锁,用更专业的话说,就是寻找药物靶标,即弄清楚药物分子作用与何种蛋白结合。如果我们拥有可解码蛋白质结构的人工智能,就可快速筛选成千上万的新药物靶标。
AlphaFold2已经预测了几乎整个人类蛋白质组的蛋白结构。由于每个蛋白都可能是某种疾病的靶点,科学家们后续可以针对每个蛋白都设计出对应的分子进行调控(抑制或者激活),这也将可能改变颠覆传统药物的研发过程,靶向单一蛋白的新药将越来越少,而靶向蛋白-蛋白相互作用的小分子或者抗体药物可能成为新药的主流。
不仅如此,上面提到的这两种强大的基于人工智能的蛋白质结构预测模型,RoseTTAFold 的代码和服务器完全免费提供给科学界,同样DeepMind创始人也在Nature杂志上分享了AlphaFold的开源代码,并发表了系统的完整方法论,详尽细致说明AlphaFold是如何做到精确预测蛋白质3D结构的。科研工作者可以随时利用这两款模型获取蛋白质的空间结构,而无需对蛋白质进行结晶或使用昂贵的冷冻电镜进行研究。相关程序已被多个独立科研团队从GitHub免费下载,来自世界各地的科学家现在正在使用这些人工智能技术来构建蛋白质模型,以加速相关领域的研究。基于人工智能预测蛋白质结构的技术也同时被Nature和Science评为年度十大科技突破。
Science 杂志公布的 2021 年年度科学突破榜单,AI预测蛋白折叠结构位列榜首。而Nature Methods 发布的年度方法专刊,也将蛋白质结构预测评为2021年的年度科技方法——这是15年来计算方法第一次被评为年度 Nature Method。

Science杂志2021十大科学突破榜首(图源:[4])
当然目前AI预测的蛋白结构仍然有很多局限。很多蛋白通过与其它蛋白、核苷酸或配体结合来行使功能,AI尚且不能预测复杂复合体的三维结构。此外,蛋白构象很多情况下是动态过程,同一个蛋白可能根据环境和其它因素,变换成不同的构象并且具有不同的功能,而人工智能通常只能预测出一个最优构象,也不能预测蛋白的动态过程。AI在蛋白质结构预测领域做出了惊人的成绩,但这并不意味着AI明白地告诉了我们蛋白质折叠的过程与原理。人工智能预测蛋白质结构这一里程碑式的进展,距离人类完全解析生命活动的奥秘仍有一段的距离。而未来,这一技术将以怎样一种方式改变生命科学,将如何加速疾病研究、药物研发等领域,让我们拭目以待!
参考资料:
[1] http://sciencewithmsjones.weebly.com/living-environment/central-dogma-of-genetics?fbclid=IwAR13Gz372IrbOBKoNi3BWheUb9gwbrCgW30oApkw1lEwl19EHLMkJ75lAzI
[2] Jumper, J. et al. Nature https://doi.org/10.1038/s41586-021-03819-2 (2021)
[3] Callaway, E Nature https://doi.org/10.1038/d41586-021-02025-4 (2021)
[4] https://www.science.org/content/article/breakthrough-2021#
[5] Tunyasuvunakool, K. et al. Nature https://doi.org/10.1038/s41586-021-03828-1 (2021)
[6] Baek, M. et al. Science https://doi.org/10.1126/science.abj8754 (2021)



 粤公网安备44030002003406
粤公网安备44030002003406